Structure of the Heme Prosthetic Group
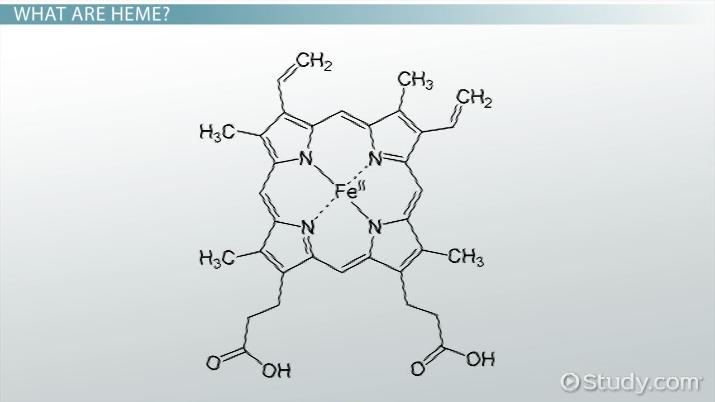
Heme is the O2 – binding molecule common to Mb and Hb protophorphyrin IX is the backbone
of heme when iron is complexed with protophorphyrin IX it is called Heme. So heme is the
prosthetic group in Hemoglobin, Myoglobin and Cytochrome b, c, and c1.
The Fe – porphyrin prosthetic group is, with the exception of two propionate groups,
hydrophobic and planar.
Heme become an integral part of the globin proteins during poly peptide synthesis. It is the
heme molecule that give globin proteins their characteristic red brown colour. Once the Fe2+
(Ferrious) is incorporated, the protein is called hemoglobin. Such structural coordination creates
an environment essential for Globin to bind and release O2.
If the iron atom were to become oxidized to Fe3+(Ferric), the Globin would get changed to
metmyoglobin or (Met hemoglobin) where heme can no longer interact with O2 and O2 transport
is compromised.

Fig 5.21: Structure of the heme prosthetic group (protophorphyrin IX) ring system.
Heme is non-covalently bonded in a hydrophobic crevice in the myoglobin and hemoglobin
molecules.
Ferrous iron is octahedrally coordinated having six ligands or binding groups, attached to it,
the nitrogen atoms account for only four ligands. The two remaining coordination sites which
lie along the ring contain on the plane of the ring contains one histidine with imidazole
nitrogen that is close enough to bond directly to the Fe2+ called proximal histidine the other
histidine which facilitates the alignment of heme to O2 and that of Fe2+ called distal
Histidine.Distal histidine confers important geometrical constraints on the six coordination
sites which normally restricts the interaction with CO.
The coordinate nitrogen atoms mainly prevents conversion of the heme iron to the ferric state
(Fe3+) due to their electron donating character.
In free heme molecules, reaction of oxygen at one of the two “open” coordination bonds of iron
which is perpendicular to the plane of the porphryin molecule above and below can result in
irreversible conversion of Fe2+ to Fe3+. In heme containing proteins this reaction is prevented by sequestering the heme deep within a protein structure where access to the two open
coordination bonds is restricted polar amino acids are located almost exclusively on the exterior
surface of globin polypeptide and contribute to the high solubility of these proteins. Amino acids
which are both polar and hydrophobic, such as Threonine, tyrosine and Tryptophan are oriented
to the exterior.
Hydrophobic amino acid residues are buried with in the interior where they stabilize the folding
of the polypeptide and binding of iron porphyrin ring.
The only exceptions to this general distribution of amino acids residues in globins are the two
Histidines that play an indispensable role in the heme binding are oriented perpendicular to and
on either side of the planor heme prosthetic group.
In the quaternary stucture of human Hb there exists two α- globin and two – β- globin sub units
(α2 β2). These subunits are arranged in a tetrahedral array. Experimental analysis of the
quaternary structure indicates multiple non-convalent interactions between each pair of
dissimilar subunits, that is, at the α - β - interfaces. In contrast there are few interactions
between identical subunits at the α - α or β – β interface so hemoglobin is considered more as a
heterodmer (α β)2. The α β -heterodimer are now recognized as major factors determiners of
O2 binding and release.
Myoglobin and Hemoglobin
Both Myoglobin and Hemoglobin are built on a common structural motif. Myoglobin contains a
single polypeptide chain folded about a prosthetic group, the heme, which contains the oxygen
binding site. Hemoglobin is a tetrameric protein. Each polypeptide subunit closely resembles
myoglobin. Note, for example that myoglobin and each subunit of hemoglobin consists of eight
helical segments, which are labeled A through H. The multiple subunit structure of hemoglobin
gives it important oxygen binding properties that are different than myoglobin's, consistent with
hemoglobin's role in oxygen transport. In all vertebrates the oxygen transport protein is
hemoglobin, a protein that can pick up oxygen in lungs or gills and deliver it to tissues.
Myoglobin, by contrast, is an oxygen storage protein. Oxygen transported to tissues must be
released for utilization. In tissues, such as muscle, with high oxygen demands, myoglobin
provides large oxygen reserves.
Adult Hb (HbA)
Contains two types of globin two α - chains (141 residues each) and two β - chains (146 residue
each). The amino acid sequences of the two type of subunits are identical at 27 positions.
Fetal Hb (HbF)
Contains a different type of Hb just after conception fetuses synthesize zeta chain (quite like α -
chain)
The HbF variant barely detectable and ε- chains just like β - chain later zeta replaced by α - and
ε- by γ. HbF contain 2 γ and 2 γ subunits in most adult often increases up to 15 - 20% in
individuals with mutant adult Hbs, such as sickle cell disease. This is an example of the body’s
compensatory response to a pathologic abnormality .The direct benefit of this structural change
in Hb isoform is a more efficient transfer of O2 from maternal HbA to fetal( HbF) .
Sickle Cell Hemoglobin (HbS)
HbS, the variant most commonly associated with sickle cell disease, cannot tolerate high protein
concentration when deoxygenated. At low oxygen concentrations, deoxy HbS polymerizes,
forms fibers, and distorts erythrocytes in to sickle shapes.
The mutation is Glu6 β -val a surface localized charged amino acids is replaced by a
hydrophobic residue as show below
HbA = Val – His – Leu – Thr – Pro – Glu – Glu – Lys
HbS = Val – His – Leu – Thr – Pro – Val – Glu – Lys
Such substitution of Valine (non - polar) for Glutamate (polar) have the following consequence
1. Place A non - polar residue on the outside of HbS which markedly reduce solubility of
deoxy HbS. But has little effect on oxy - HbS (causes Hb to clump when deoxygenated)
2. Creates sticky patches on the outside surface of each β - chains (not present HbA)
3. The sticky patches interact with complementary sites of another HbS (oxy) and forms
large aggregates that distort the whole RBC structure.
Sickle Cell Trait
The heterozygote individuals (sickle cell trait) (HbA/HbS) is associated with increased
resistance to malaria. Specifically growth of the infectious agent, Plasmodium falciparum in the
erythrocyte.
This observation represents an example of a selective advantage that HbA/ HbS heterozygote
exhibits over the HbA/HbA normal or the HbS/HbS homozygte.
Sickled erythrocyte exhibits little or less deformity, they no longer move freely through the
micorvasculature and often block blood flow. Moreover this cells lose water, become fragile and
have a considerably short life span leading to anemia.
Sickle Cell Disease
Sickle cell disease is caused by an inherited structural abnormality in the β –globin polypeptide.
Clinically, an individual with sickle cell disease present with intermittent episode of haemolytic
and painful vaso–occlusive crisis. The latter leading to severe pain in bone chest and abdomen.
There is also a likely to be impaired growth, increased susceptibility to infections and multiple
organ damage.
Digestion and Absorption of Proteins
Proteins are larger polypeptide molecules coiled by weaker bonds in their tertiary structure the
digestion of proteins involves the gradual breakdown of this polypeptide by enzymatic hydrolysis
in to amino acid molecules which are absorbed in the blood stream. The protein load received
by the gut is derived from two sources 70-100g dietary protein which is required daily and 35 -
200g endogenous protein (secreted enzymes and proteins in the gut or from intestinal epithelia
cell turnover)
Only 1-2g of nitrogen equivalent to 6-12g of proteins are lost in the feces on a daily basis. Thus
the digestion and absorption of protein is more efficient.
The process of protein digestion can be divided, depending on the sources of peptidases.
A. Gastric Digestion
Entry of a protein in to stomach stimulates the gastric mucosa to secrete a hormone gastrin
which in turn stimulates the secretion of Hcl by the parietal cells of the gastric glands and
pepsinogen by the chief cells.
The HCL thus produced lower the pH of stomach to (pH1.5 – 2.5) and acts as an antiseptic and
kills most of the bacteria and other foreign cells ingested along with.
The acid denatures the protein and the whole protein susceptible to hydrolysis by the action
other proteolytic enzymes.
Proteases are endopeptidases which attack the internal bonds and liberate large fragments of
peptides.
Then pepsinogen having MW 40,000 an inactive precursor or zymogen is converted in to active
pepsin in the stomach itself. In this process 44 amino acids gets removed from the amino
terminal end and the portion of the molecule that remain intact is enzymatically active pepsin
(MW. 33,000).
This active pepsin cleaves the ingested protein at their amino terminus of aromatic amino acids
(Phe, Tyr, and Trp.)
The major products of pepsin action are large peptide fragments and some free amino acids.
B. Pancreatic Digestion
Pancreatic zymogens proceed digestion as the acidic stomach contents pass in to the small
intestine, A low pH triggers the secretion of a hormone Secretin in the blood.
Secretin stimulates the pancreas to secrete HCO3 (bicarbonate), which in the small intestine
neutralizes the gastric HCL and abruptly change the pH to 7.0.
The entry of large peptide fragments and some free amino acids in the upper part of the small
intestine (Duodenum), excites the release of a hormone cholecytokinin (CCK).
CCK:
1) stimulates gall bladder contraction.
2) stimulate secretion of several pancreatic enzymes whose activity is between pH 7and
8 in proenzyme forms.
Three of these pro-enzyme are trypsinogen, chymotrypsinogen and procarboxy peptidase,
localized in the exocrine cells.Synthesis of these enzymes as inactive precursors protects the
exocrine cells from destructive proteolytic attack.
When the proenzyme reach the lumen of the small intestine, initially the enteropeptidase (old
name Enterokinase) a protease produced by duodenal epithelial cells, activates pancreatic
trypsinogen to trypsin by the removal of a hexapeptide from NH2 – terminus
Trypsin in turn auto catalytically activates more trypsinogen to trypsin and other proenzymes
and liberating chymotrypsin elastas, and carboxypeptidase’s A and B as shown below.

By the sequential action of these proteolytic enzymes and peptides ingested proteins are
hydrolyzed to yield a mixture of free amino acids which can be transported across the epithelial
lining of the small intestine.

Table 5.4: Digestive enzymes and their specificity
C. Intestinal Digestion
Since pancreatic juice does not contain appreciable aminopeptidase activity final digestion of di
and Oligopeptides depends on the small intestinal enzymes.
The lumenal surface of epithelial cells is rich in endopeptidase, and dipptidase aminopeptidase
activity
The end products of the cell surface digestion are free amino acids and di and tripeptides.
These are passed in to the interior of the epithelial cell where other specific peptidases convert
almost all of them to a single amino acids that are transported to the blood stream by the
opposite side of the cell membrane and carried to liver (primarily) and other tissues for oxidative
degradation. This process complete the absorption of 99% of digested proteins. The whole
scheme is as shown in the figure below.

Fig 5.22: Digestion and absorption of proteins
II. Transport of Amino Acids in to Intestinal Epithelial cells.
The mechanism of active transport of amino acids are similar with that of glucose uptake.
At the brush - border membrane Na+
- dependent symporters for amino acid uptake are
functional with consequent ATP - linked pumping out Na+
at the contraluminal membrane. This
is an indirect active transport.
A similar H+ dependent symport is present on the brush border surface of di and tripeptides
active transport in to the cell.
Na+ - independent transporters are present on the contralumenal surface, thus allowing amino
acids? Facilitated transport to the hepatic portal system.
From both genetic and transporters studies at least six specific symporter systems have been
identified for the uptake of L-amino acids from the intestinal lumen.
1. Neutral amino acid symporters with short or polar side chains.
Ser, Thr, Ala,
2. Neutral amino acid symporter for aromatic or hydrophobic side chains.
Phe, Tyr,
3. lmino acid symporter Pro,and OH – Pro
4. Basic amino acid symporter Lys, Arg and Cys.
5. Acidic amino acid symporter. Asp, Glu
6. β amino acid symporter β-Ala , Tau.
These transporter systems are also present in the renal tubules and defects in their constituent
protein structure can lead to disease called Hartnup disease.
Neutral amino Aciduria (Hartnup Disease)
Transport functions, like enzymatic functions, are subject to modification by mutations. An
example of a genetic lesion in epithelial amino acid transport is hartnup disease; entry resulting
from the defect was first recognized. The disease is characterized by the inability of renal and
intestinal epithelial cells to absorb neutral amino acids from the lumen. In the kidney, in which
plasma amino acids reach the lumen of the proximal tubule through the Ultra filtrate, the inability
to reabsorb amino acids manifests itself as excretion of amino acids in the Urine
(aminoaciduria). The intestinal defect results in malabsorption of free amino acids from the diet.
Therefore the clinical symptoms of patients with this are mainly those due to essential amino
acid and Nicotinamide deficiencies. The pellagra-like features are explained by a deficiency of
Tryptophan, which serves as precursor for nicotinamide. Investigations of patients with Hartnup
disease revealed the existence of intestinal transport systems for di - or tripeptides, which are
different from the ones for free amino acids. The genetic lesion does not affect transport of
peptides, which remains as a pathway for absorption of protein digestion products.